在做比较基因组学分析的时候,经常会遇到一种需求:我们想看某一个基因在不同物种中的分布情况及基因结构的区别。
在很多文献中比较常见的一种思路是:通过一个query序列在不同物种中进行blast搜索,获取基因组上大致的范围,然后用Genewise进行基因预测,如果是新测的物种,那么由于NCBI上没有这一物种的注释结果,我们只能这么做。但是如果我们要在已经发表的基因组中看某一个基因的情况的话,最好先在NCBI数据库中看下这个基因是否在这些基因组中已经注释出来。
因为无论我们自己的注释始终比不上NCBI的注释方法全面,因为NCBI的注释综合了NCBI中大量的数据,例如EST、cDNA等等,我们自己去注释的话没有这么多数据来源,所以对于
已经发表的基因组注释,我们的注释效果肯定比不上NCBI自己注释的效果。
比方说,如果我们要看CYP2J19这个基因在所有的已经发表的鸟类基因组中的分布情况,那么首先去NCBI上搜索这一基因:
由于鸡是鸟类中比较有代表性的物种,我们以鸡当中的这个基因作为query,点击右侧的Run Blast,在Entrez Query中输入biomol_mrna[properties] AND avian[organism],意思是只需要mRNA序列,物种范围为鸟纲,具体可参考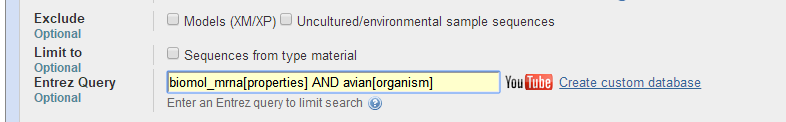
在下方的参数设置中设置为最多250个结果,以得到尽量多的结果: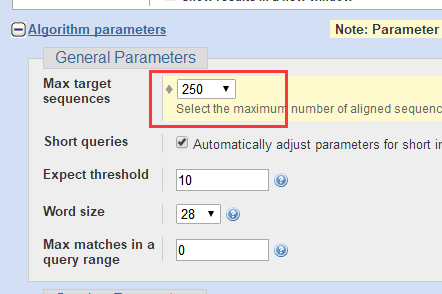
点击BLAST,登上差不多1分钟,NCBI就有结果了。然后选择All:
点击下载完整序列(这样我们会得到这些匹配上结果的完整序列,而不止是比对上的部分):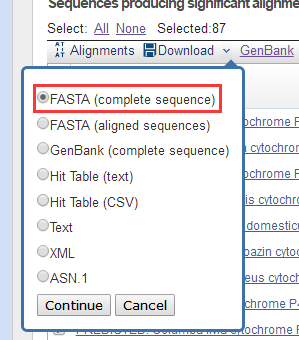
但是这样一来就有了第二个问题:NCBI上的基因序列分为mRNA和cds,两者的区别在与,mRNA序列前后经常会有UTR片段,而cds序列不包括UTR序列,因此cds才是蛋白序列所对应的核酸序列,也就是说cds经过翻译后可以成为蛋白序列。刚才的下载选项里只有完整序列即mRNA序列,我们现在如果只想获得cds序列的话,该怎么办?
我这里有一个曲线救国的方法:
先获得这些序列的编号,然后通过Batch Entrez批量获得cds序列。
获得序列编号(其中seqdump.txt是刚刚下载的mRNA序列文件)1
grep ">" seqdump.txt|awk '{print $1}'|awk -F">" '{print $2}' >download.list
然后打开Batch Entrez,
数据库选择Nucleotide,然后选择文件上传,再点击右侧的Retrieve,出现如下界面: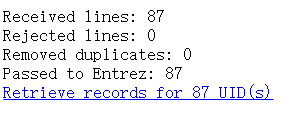
然后点击Retrieve records for 87 UID(s),然后看到批量的结果: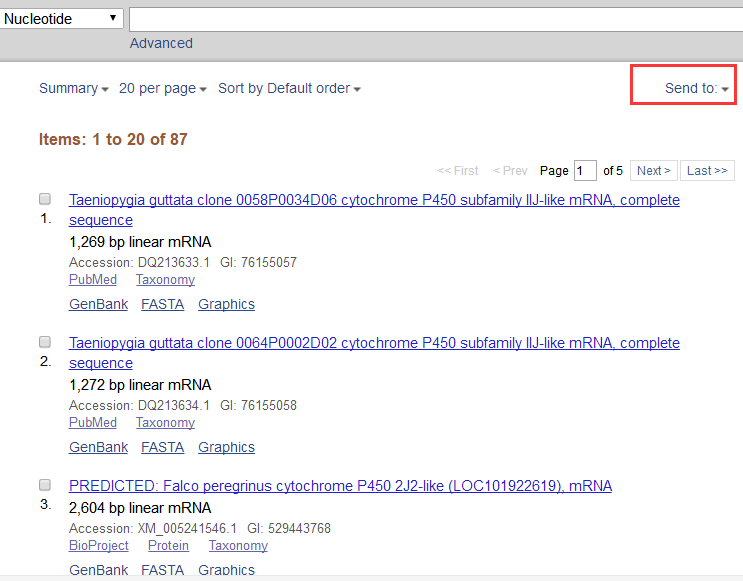
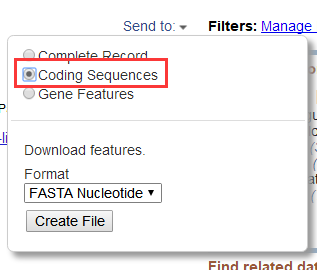
这时点击右上角的Send to,选择Coding Sequences,再点击Creat File,即可获得文件,